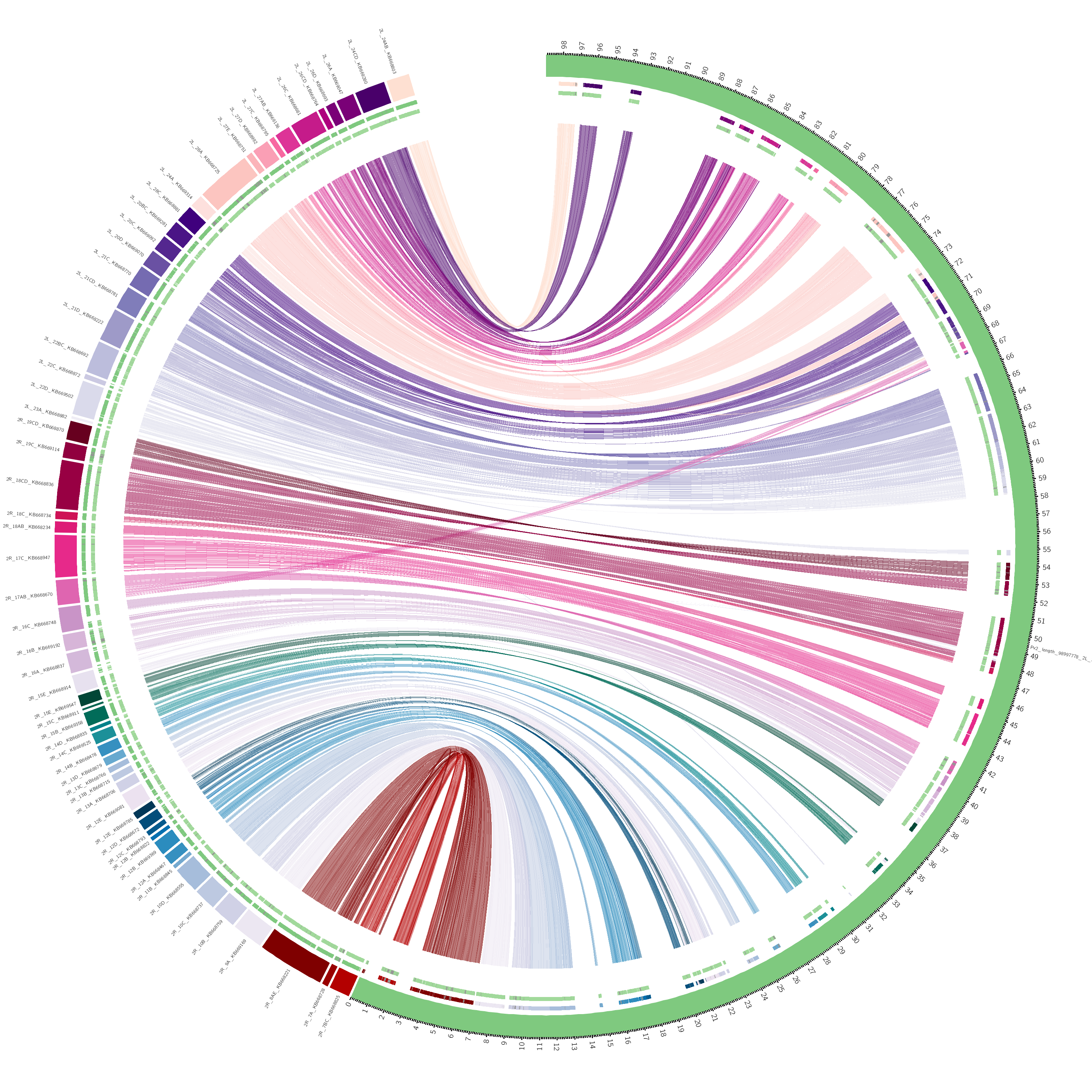
 Fig. 1. Pairwise alignment of the 2015 An funestus Fumoz assembly (left) and the improved An funestus Fumoz assembly (right) for Chromosome 2. Note that the large number of contigs in the 2015 assembly align with few rearrangements to the improved assembly. This assures that there are few chimeric mis-assemblies.
Fig. 1. Pairwise alignment of the 2015 An funestus Fumoz assembly (left) and the improved An funestus Fumoz assembly (right) for Chromosome 2. Note that the large number of contigs in the 2015 assembly align with few rearrangements to the improved assembly. This assures that there are few chimeric mis-assemblies.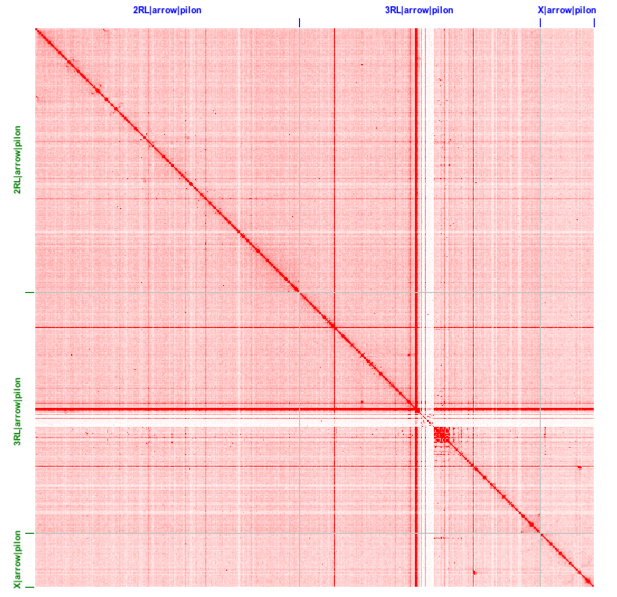 Fig. 2. Heatmap of HiC data mapped to the 3 scaffolds (chromosomes), indicating interaction between all 5k non-overlapping windows in the genome.
Fig. 2. Heatmap of HiC data mapped to the 3 scaffolds (chromosomes), indicating interaction between all 5k non-overlapping windows in the genome. Anchoring and uniting the An. funestus assembly for improved vector analysis
Anopheles funestus is one of the three most important and widespread vectors of human malaria in tropical Africa, but unlike An. gambiae with which it broadly co-occurs, it is a relatively neglected species. It shares with An. gambiae not only a broad sub-Saharan distribution and major vector status, but also abundant chromosomal inversion polymorphism and shallow population structure across much of Africa. However, there are behavioral and genetic heterogeneities relevant to malaria transmission that remain poorly understood. In the savannas of West Africa, where application of residual insecticides in the 1960's was not as successful against An. funestus as elsewhere in Africa, there is strong cytogenetic evidence for cryptic, temporally stable assortatively mating populations co-occurring in the same villages. In apparent analogy to the chromosomal forms of An. gambiae, the chromosomally recognized forms of An. funestus, named Kiribina and Folonzo, differ in larval ecology. Importantly, they also differ in adult behaviors affecting vectorial capacity, most notably indoor/outdoor resting behavior. At present, there exist no rapid molecular identification assays to facilitate more in-depth field studies of their behavior and genetics; the forms can only be distinguished by laborious chromosomal karyotyping.
Our long-term goal is to understand the underlying genomic determinants of epidemiologically important phenotypic and behavioral traits in An. funestus and its chromosomal forms. The reference An. funestus genome assembly from 2015 begins to make possible this goal, but its fragmented state, unanchored to chromosomes, poses a barrier that hinders the identification of causal loci affecting traits of interest. We therefore set about to upgrade the draft An. funestus reference to a chromosome-based contiguous assembly using extensive long single molecule sequencing (PacBio) and Hi-C libraries from the same FuMoz strain. Using the resulting assembly, we are conducting an assessment of genomic divergence between ~190 cytogenetically karyotyped and individually whole genome-sequenced Folonzo and Kiribina mosquitoes from Burkina Faso.
Funding: NIH R21 AI112734
References:
Collaborators (Past and Present):